Developing a new medicine is a long and rigorous process, and much of what tested and tried never makes it to patients. In the search for new medicines, the primary concern is patient safety.
On average it takes about 12-15 years to progress from a promising idea in the research laboratory to receiving approval from regulators. Clinical development is the most time intensive and expensive part of this research and development continuum accounting for approximately 45 to 75 percent of the USD 1.2 billion average cost of bringing a new therapy to market.
Developing a new treatment typically goes through the following stages from laboratory to patient:
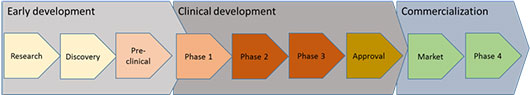
Early development
Research – identifying a new approach to treat a disease or condition
The pharma industry uses the growing understanding of biology to identify ‘targets’. A target is a structure – like an enzyme – in the body that contributes to the development of a disease or its symptoms. It is important initially to know that the selected target is valid, and that it affects the disease process in the way that the science suggests. Once a target has been identified, the next step is to find chemicals or natural compounds that might inhibit or enhance the target’s activity. Current techniques allow many hundreds of compounds to be screened for that sort of activity very quickly. Compounds that show desired effects are called ‘hits’.
Discovery – developing and investigating compounds that can alter a disease process
Once the hits are identified, the number of possible compounds are narrowed down by further testing, to come up with lead compounds. A lead compounds may need to be ‘improved’ – it may be modified by using chemistry to enhance different properties. Ideally we look for compounds that:
- selectively hit the target for the disease processes
- can be safely absorbed by the body
- reach the appropriate point in the body to find the target
- remain in the system long enough to have an effect
- can be manufactured to produce a stable preparation convenient to use
- have few side effects
Pre-clinical testing – understanding how a compound works in a system and its effects
At this stage researchers do more detailed investigation of the lead compounds. By testing with living cells, bacteria or tissue cultures in the lab, or in animals, scientists can build an understanding of how the compounds behave.
If the work has been successful, the development team will have identified a compound to consider testing in humans.
Clinical development
Clinical trials – carrying out tests in humans
Before any trial in humans can start, a plan is developed that defines what the trial would involve. The plan will be carefully considered by an ethical review committee of experienced independent medical and scientific practitioners. This committee has the power to reject or stop a clinical trial at any time.
New medicines must go through three phases of human testing before they can be approved for use.
The safety of those who take part in clinical trials is of paramount importance. All our clinical trials follow the guidelines developed by the International Conference on Harmonisation (ICH), and the World Medical Association Declaration of Helsinki on the Ethical Principles for Medical Research Involving Human Subjects (2008).
Phase 1
Phase 1 clinical trials mark the first time an experimental therapy is administered to humans. Phase 1 trials usually focus on ensuring the therapy is safe to use in people, rather than how effective it may be as a treatment for a given disease.
During this phase, escalating doses of the experimental therapy are given to a small number of study participants so that researchers can measure the body’s response, including how it is absorbed, its duration in the blood stream and which dosage levels are safe and well tolerated.
Phase 2
Phase 2 trials – also called ‘proof of concept’ studies – generally assess the effectiveness of an experimental therapy at treating a specific illness or medical condition. Information about the experimental therapy’s safety, side effects and potential risks are also collected. In this phase, researchers work to determine the most effective dosages for the experimental therapy and the most appropriate method of delivery, such as tablets, extended release capsules, infusions or injections.
Phase 2 clinical trials involve a larger number of trial participants, typically up to several hundred, who usually have the medical condition that the experimental therapy is intended to treat.
Phase 3
Phase 3 trials test the results of earlier trials in much larger groups of people and gather additional information on the effectiveness and safety of the experimental therapy. This phase will usually involve several hundred to several thousand participants across multiple study locations. These trials are often randomized, where participants are arbitrarily allocated to receive the experimental therapy, placebo or another therapy (a “comparator”), and “double-blinded,” in which neither the investigator nor the participant are aware if the therapy given is the true experimental therapy, placebo (medication with no active ingredients) or another therapy (a “comparator”).
Phase 3 trials generally provide the primary basis for the benefit-risk assessment for the new therapy and much of the core information about the therapy that is analyzed for inclusion in final labeling if approved by the regulatory authority.
Registration
Filing an application for registration with the country’s health regulatory authority is the next step in bringing a potential new therapy to patients. In the U.S., a New Drug Application (NDA) is filed with the U.S. Food and Drug Administration (FDA). In Europe, a Market Authorization Application (MAA) is filed with the European Agency for the Evaluation of Medicinal Products (EMEA). A description of the therapy’s manufacturing process along with quality data and trial results from all pre-clinical and clinical trials are provided in order to demonstrate the safety and effectiveness of the new therapy.
If approval is granted, the new therapy can then be sold for use as detailed in the final approval label granted by the regulatory authority.
Commercialization
Market
Once the drug is approved for marketing it can be launched and promoted to target prescriber and patient groups. Marketing, sales and distribution of drug are strictly regulated activities. This to safeguard proper and safe use of the drug.
Phase 4
Phase 4 trials — also called “post-marketing studies” — are conducted after regulatory approval and are critical to informing the ongoing use of the therapy. Through such trials, researchers collect additional information about longer-term risks, benefits and optimal use. These trials often involve thousands of participants and may continue for many years.